


 Sign Out
Sign Out

Paracetamol is another centrally acting analgesic. The exact site and mechanism of its analgesic action is not clearly defined.
When evaluated in a standard animal model, the combination of tramadol and paracetamol exhibited a synergistic effect.
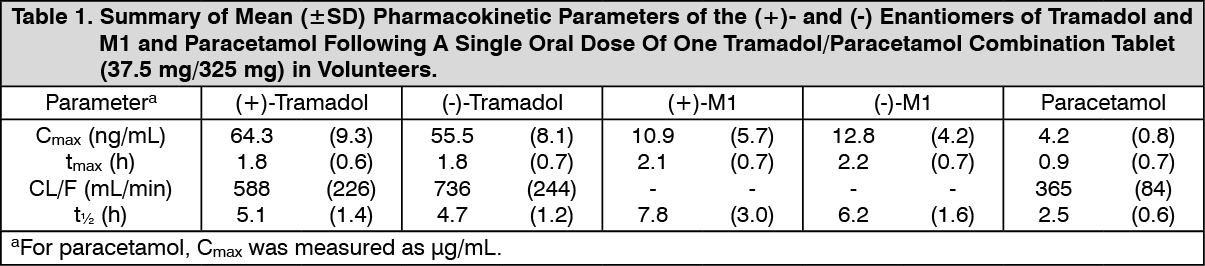
Pharmacokinetics: General: Tramadol is administered as a racemate and both the [-] and [+] forms of both tramadol and M1 are detected in the circulation. The pharmacokinetics of plasma tramadol and paracetamol following oral administration of one tramadol/paracetamol (Dolcet Mini) tablet are shown in Table 1. Tramadol has a slower absorption and longer half-life when compared to paracetamol.
After a single oral dose of one Tramadol/Paracetamol combination tablet (37.5 mg/325 mg) peak plasma concentrations of 64.3/55.5 ng/ml [(+)-Tramadol/(-)-Tramadol] and 4.2 μg/ml (paracetamol) are reached after 1.8 h [(+)-Tramadol/(-)-Tramadol] and 0.9 h (paracetamol), respectively. Mean elimination half lives (t1/2) are 5.1/4.7 h [(+)-Tramadol/(-)-Tramadol] and 2.5 h (paracetamol).
Single and multiple dose pharmacokinetic studies of tramadol/paracetamol (Dolcet Mini) in volunteers showed no significant drug interactions between tramadol and paracetamol. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAbsorption: Tramadol hydrochloride has a mean absolute bioavailability of approximately 75% following administration of a single 100 mg oral dose of tramadol tablets. The mean peak plasma concentration of racemic tramadol and M1 after administration of two tramadol/paracetamol (Dolcet Mini) tablets occurs at approximately two and three hours, respectively, post-dose in healthy adults.
Oral absorption of paracetamol following administration of tramadol/paracetamol (Dolcet Mini) is rapid and almost complete and occurs primarily in the small intestine. Peak plasma concentrations of paracetamol occur within 1 hour and are not affected by co-administration with tramadol.
Food effects: The oral administration of tramadol/paracetamol (Dolcet Mini) with food has no significant effect on the peak plasma concentration or extent of absorption of either tramadol or paracetamol, so that tramadol/paracetamol (Dolcet Mini) can be taken independently of meal times.
Distribution: The volume of distribution of tramadol was 2.6 and 2.9 L/kg in male and female subjects, respectively, following a 100 mg intravenous dose. The binding of tramadol to human plasma proteins is approximately 20%.
Paracetamol appears to be widely distributed throughout most body tissues except fat. Its apparent volume of distribution is about 0.9 L/kg.
A relative small portion (~20%) of paracetamol is bound to plasma protein.
Metabolism: Plasma concentration profiles for tramadol and its M1 metabolite measured following dosing of tramadol/paracetamol (Dolcet Mini) in volunteers showed no significant change compared to dosing with tramadol alone.
Approximately 30% of the dose is excreted in the urine as unchanged drug, whereas 60% of the dose is excreted as metabolites. The major metabolic pathways appear to be N- and O-demethylation and glucuronidation or sulfation in the liver. Tramadol is extensively metabolized by a number of pathways, including CYP2D6. Patients who are CYP2D6 ultra-rapid metabolizers may convert tramadol to its active metabolite (M1) more rapidly and completely than other patients (see CYP2D6 Ultra-Rapid Metabolism of Tramadol under Precautions). The prevalence of this CYP2D6 genotype varies by population and has been reported in literature to range from 1% to 10% in African Americans, Caucasian Americans, Asians and Europeans (including specific studies in Greeks, Hungarians and Northern Europeans) to as high as 29% in African/Ethiopians.
Paracetamol is primarily metabolized in the liver by first-order kinetics and involves three principle separate pathways: a) conjugation with glucuronide; b) conjugation with sulfate; and c) oxidation via cytochrome P450 enzyme pathway.
Excretion: Tramadol and its metabolites are eliminated primarily by the kidney. The plasma elimination half-lives of racemic tramadol and M1 are approximately six and seven hours, respectively. The plasma elimination half-life of racemic tramadol increased from approximately six hours to seven hours upon multiple dosing of tramadol/paracetamol (Dolcet Mini).
The half-life of paracetamol is about 2 to 3 hours in adults. It is somewhat shorter in children and somewhat longer in neonates and in cirrhotic patients. Paracetamol is eliminated from the body primarily by formation of glucuronide and sulfate conjugates in a dose-dependent manner. Less than 9% of paracetamol is excreted unchanged in the urine.
Toxicology: Non-Clinical Information: Tramadol/Paracetamol combination: There are no animal or laboratory studies on the combination product (tramadol and paracetamol) to evaluate carcinogenesis, mutagenesis, or impairment of fertility.
No drug-related teratogenic effects were observed in the progeny of rats treated orally with the combination of tramadol and paracetamol. The tramadol/paracetamol combination product was shown to be embryotoxic and fetotoxic in rats at a maternally toxic dose (50/434 mg/kg tramadol/paracetamol) 8.3 times the maximum human dose but was not teratogenic at this dose level. Embryo and fetal toxicity consisted of decreased fetal weights and increased supernumerary ribs. Lower and less severe maternally toxic dosages (10/87 and 25/217 mg/kg tramadol/paracetamol) did not produce embryo or fetal toxicity.
Tramadol hydrochloride: Carcinogenicity/Mutagenicity: A slight but statistically significant increase in two common murine tumors, pulmonary and hepatic, was observed in a mouse carcinogenicity study, particularly in aged mice (dosing orally up to 30 mg/kg for approximately two years, although the study was not done with the Maximum Tolerated Dose). This finding is not believed to suggest risk in humans. No such finding occurred in a rat carcinogenicity study.
Tramadol was not mutagenic in the following assays: Ames Salmonella microsomal activation test, CHO/HPRT mammalian cell assay, mouse lymphoma assay (in the absence of metabolic activation), dominant lethal mutation tests in mice, chromosome aberration test in Chinese hamsters, and bone marrow micronucleus tests in mice and Chinese hamsters.
Weakly mutagenic results occurred in the presence of metabolic activation in the mouse lymphoma assay and micronucleus test in rats. Overall, the weight of evidence from these tests indicates that tramadol does not pose a genotoxic risk to humans.
Fertility: No effects on fertility were observed for tramadol at oral dose levels up to 50 mg/kg in male rats and 75 mg/kg in female rats.
Effect on Reproduction: Tramadol was evaluated in peri- and post-natal studies in rats. Progeny of dams receiving oral (gavage) dose levels of 50 mg/kg or greater had decreased weights, and pup survival was decreased early in lactation at 80 mg/kg (6 to 10 times the maximum human dose). No toxicity was observed for progeny of dams receiving 8, 10, 20, 25, or 40 mg/kg. Maternal toxicity was observed at all dose levels of tramadol in this study, but effects on progeny were evident only at higher dose levels where maternal toxicity was more severe.
Others: Tramadol shows tolerability in animal tests, thus continuous use and dose increasing must be used with caution.
Animal and laboratory studies in order to evaluate carcinogenicity, mutagenicity and fecundability disorders of this drug were not conducted.
Carcinogenicity: In the study of carcinogenicity in mice which received tramadol 30mg/kg (90mg/m2, 0.5-fold of the maximum clinical dose per day 185mg/m2) for 2 years, there was little oncogenesis but statistically increased significantly. Risk of this oncogenesis is not considered in human and was not observed in the carcinogenesis study in rats administered with tramadol 30 mg/kg (180 mg/m2, 1-fold of the maximum clinical dose per day).
Genotoxicity: Tramadol showed negative in reverse mutation test using microorganism, chromosome abnormality test using mammal culture cells, genomic mutation test using CHO cells, direct method among mice lymphoma tk+/- genomic mutation test, micronucleus test using mice and hamsters, and dominant lethal test using mice, but induced mild variation in metabolic activity method and rat micronucleus test among mouse lymphoma genomic mutation test. Given overall test results, tramadol does not have risk of genotoxicity in human body.
Fecundability: In the test that administered 50 mg/kg (350 mg/m2, 1.6 times than the maximum daily clinical dosage of 185 mg/m2) of tramadol and 75 mg/kg (450 mg/m2, 2.4 times than the maximum daily clinical dosage of 185 mg/m2) to female rats, no effects on fecundability was observed.